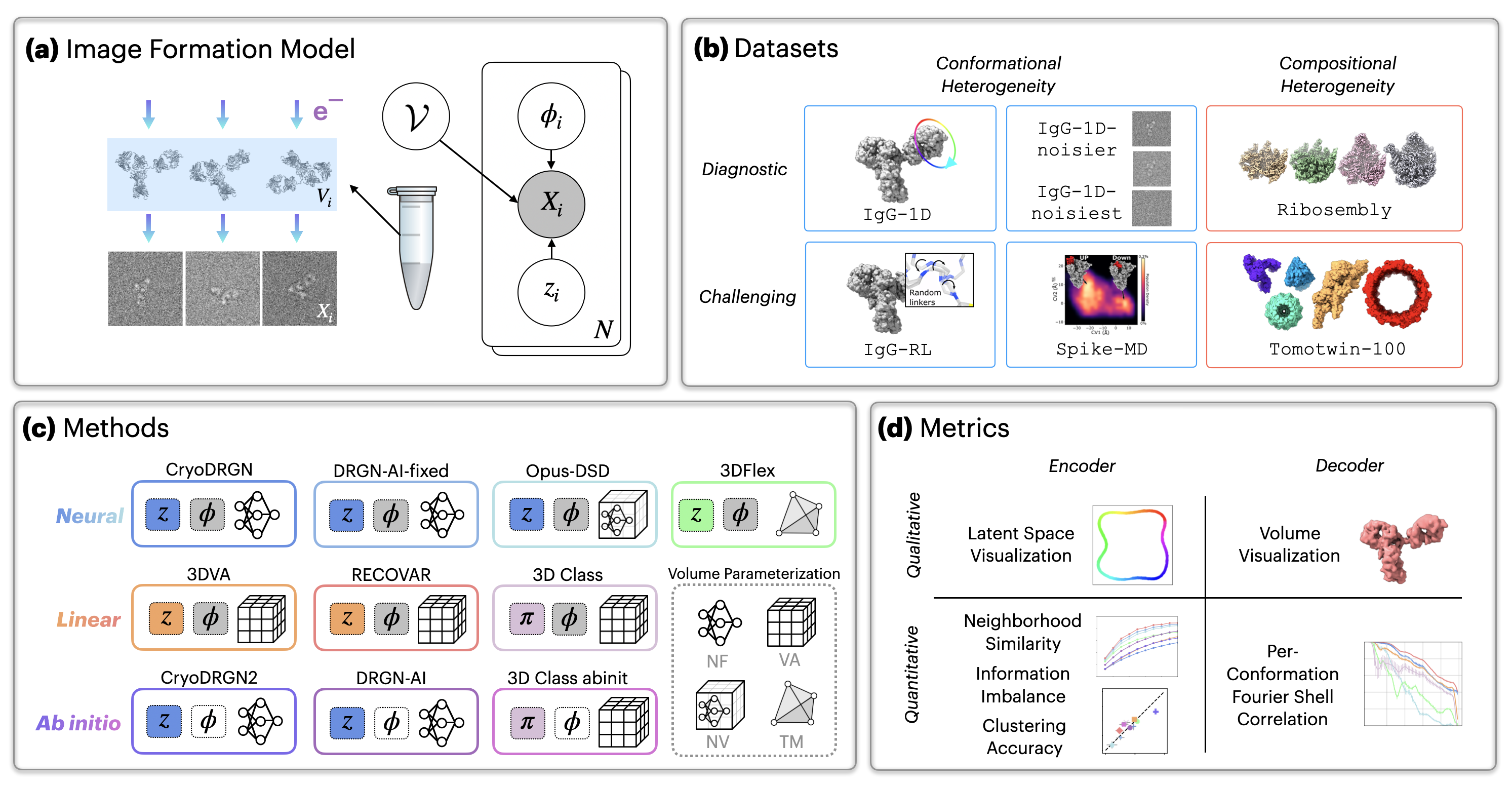
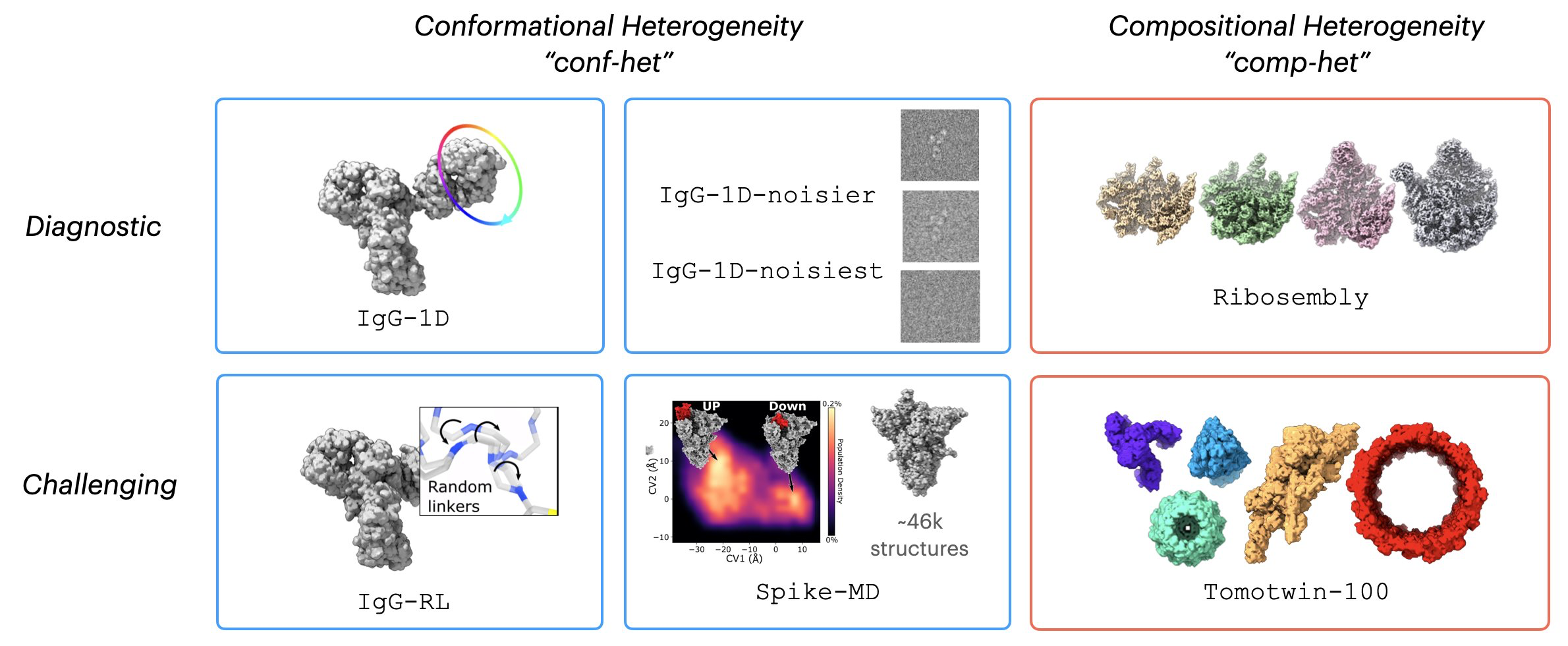
Datasets
CryoBench datasets represent both simple motions that are easy to
interpret for diagnostic purposes as well as challenging datasets to
motivate new methods development.
IgG - 1D
IgG-1D is produced by rotating one of the domains of the IgG
antibody complex 360 degrees, simulating a simple one-dimensional
continuous circular motion.
IgG - RL
For IgG-RL, we generate random conformations for a disordered
peptide linker connecting the Fab to the rest of the IgG complex.
IgG-RL is a more challenging, complex motion that hopefully
represents a realistic case of conf-het in cryo-EM.
Spike - MD
In Spike-MD, we use a long timescale molecular dynamics simulation
to produce over 46k ground truth structures in this dataset. We hope
to motivate methods development connecting MD simulations with
cryo-EM.
Ribosembly
Ribosembly provides a simple example of compositional heterogeneity
(comp-het) using 16 ribosome assembly states as ground truth
structures. These structures contain a common core that grows
through the addition of proteins and ribosomal RNA.
Tomotwin-100
Finally, Tomotwin-100 is a challenging dataset for modeling
compositional heterogeneity. It contains a mixture of 100 complexes
commonly found inside cells (h/t the TomoTwin paper for curating
these structures).
Noisy Dataset
Cryo-EM images are characteristically extremely noisy! To test the
robustness of different methods to noise, we also created the
IgG-1D-noisier and IgG-1D-noisiest dataset.
For each of these datasets, we benchmarked 10 state of the art
heterogeneous reconstruction algorithms. Check out the SI for a
detailed analysis, and please let us know if you have any feedback.